热线:400-152-6858
 显微成像
显微成像 结构分析
结构分析 成分分析
成分分析 物性分析
物性分析 热学性能
热学性能 声光电磁
声光电磁 材料加工
材料加工 原位测试
原位测试 力学性能
力学性能 环境试验
环境试验 显微成像
显微成像 病理检测
病理检测 细胞实验
细胞实验 微生物实验
微生物实验 动物实验
动物实验 测序平台
测序平台 蛋白实验
蛋白实验 分子及生化
分子及生化 分子互作平台
分子互作平台 代谢组学平台
代谢组学平台 临床前CRO
临床前CRO 蛋白组学平台
蛋白组学平台 生物信息学平台
生物信息学平台 单细胞组学
单细胞组学 整体课题服务
整体课题服务 药物筛选平台
药物筛选平台 第一性原理
第一性原理 分子动力学
分子动力学 量子化学
量子化学 CAE仿真
CAE仿真 更多服务
更多服务 超算服务
超算服务 原位变温/气氛/光照测试
原位变温/气氛/光照测试 原位电化学测试
原位电化学测试 原位力学测试
原位力学测试 吸收
吸收 散射/衍射
散射/衍射 原位
原位 成像
成像 光谱
光谱 微区
微区 论文翻译
论文翻译 数据分析
数据分析 基金标书
基金标书 文库百科
文库百科第一性原理吸附能计算的基本原理及应用领域分析
来源: 时间:2023-03-15 16:17:17 浏览:5425次
第一性原理吸附能计算是一种基于量子力学原理的计算方法,用于预测材料表面或界面上分子吸附的能力。它可以在没有实验数据的情况下预测材料的吸附性能,是材料科学和化学领域中的重要工具。在本文中,测试狗将解释第一性原理吸附能计算的基本原理、计算流程和应用。

第一性原理吸附能计算的基本原理是基于密度泛函理论(DFT)。DFT是一种计算方法,用于解决分子的量子力学问题。DFT利用电子密度来描述物质中的电子分布,通过解决薛定谔方程来预测物质的性质。在DFT中,分子中的每个原子都被视为一个带电的球,其电荷分布由其电子密度确定。通过解决薛定谔方程,可以确定每个原子周围的电子密度,从而计算出整个分子的电子密度。由此,可以得到分子的各种性质,如能量、电荷密度等。
第一性原理吸附能计算的基本步骤包括:
1.选择材料:首先需要选择要计算吸附能的材料。
2.构建模型:利用软件构建模型,包括材料的结构和分子的结构。
3.计算电子结构:通过DFT计算分子在材料表面吸附的能量。
4.分析结果:分析计算结果,以预测分子在材料表面吸附的能力。
第一性原理吸附能计算可以用于各种应用,例如材料设计、催化剂开发、气体分离和传感器等。例如,在催化剂开发中,可以通过计算不同催化剂表面的吸附能来预测其催化反应活性。在气体分离中,可以计算不同材料表面对不同气体的吸附能,以设计更有效的气体分离材料。在传感器应用中,可以计算分子在传感器表面的吸附能,以评估传感器的灵敏度和选择性。
总之,第一性原理吸附能计算是一种基于量子力学原理的计算方法,可以用于预测材料表面或界面上分子吸附的能力。通过计算分子在材料表面吸附的能量,可以预测材料的吸附性能,是材料科学和化学领域中的重要工具。使用第一性原理吸附能计算,可以预测各种分子在材料表面的吸附能,从而预测其在材料中的吸附行为和反应活性。这有助于设计和开发新的材料和催化剂,并优化其性能。
在第一性原理吸附能计算中,材料表面的电子结构是一个关键因素。材料表面的原子和分子有不同的化学反应活性,这取决于其表面的电子结构。因此,在计算吸附能时,必须考虑材料表面的电子结构。材料表面的电子结构可以通过计算材料表面的晶体结构和电子结构来获得。
另一个重要的因素是分子的电子结构。分子的电子结构决定了其在材料表面的吸附能。例如,具有不饱和化学键的分子具有更强的吸附能力,因为它们能够与表面原子形成更强的化学键。
在第一性原理吸附能计算中,计算方法的选择也非常重要。有许多不同的计算方法可用于计算吸附能,包括密度泛函理论(DFT)、量子力学分子动力学(QMMD)和紧束缚法(Tight-binding)等。每种方法都有其优缺点,选择适当的方法可以提高计算的准确性和效率。
需要注意的是,第一性原理吸附能计算也存在一些限制。由于计算成本较高,它通常只适用于研究小分子在材料表面的吸附能力。对于较大的分子和复杂的反应体系,它的计算复杂性会大大增加。此外,计算结果也可能受到实验误差、近似和计算参数的影响,因此需要谨慎分析和评估。
相关文章
一文详解扫描电子显微镜(SEM)的工作原理及应用技术
2023-10-08
接触角测试(CA)的原理、样品制备要求及实际应用
2023-11-16
热重分析(TG-DTG)曲线的几种解析方法
2023-12-26
恒电流间歇滴定法GITT的基本原理以及测试教程
2022-08-12
TMA技术的基本概念、应用领域以及实际操作中的要点
2023-12-06
一文详细介绍he染色的基本原理、实验步骤及注意事项
2023-11-23
项目推荐/Project
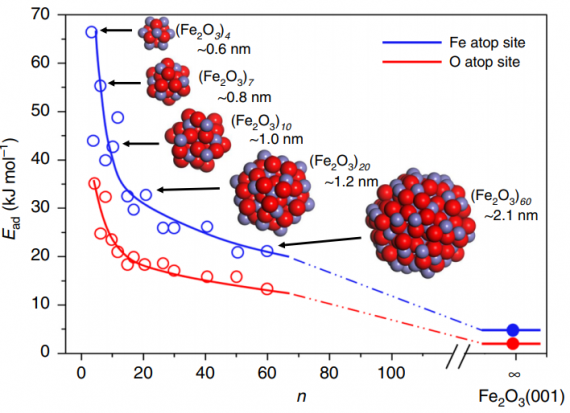
第一性原理-吸附能
热门文章/popular
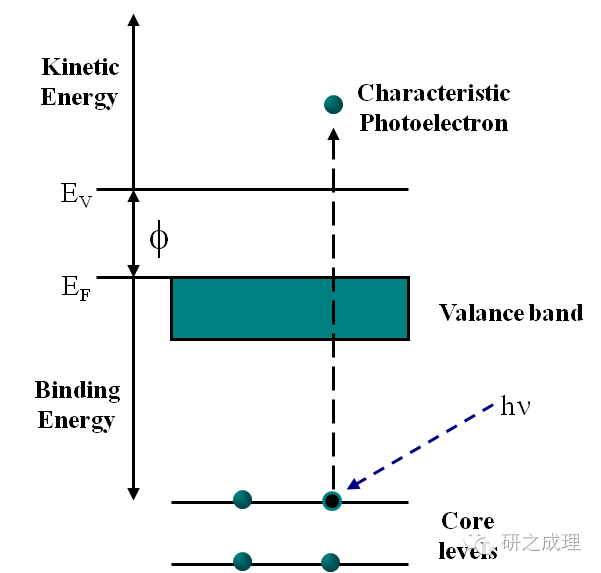
基础理论丨一文了解XPS(概念、定性定量分析、分析方法、谱线结构)

手把手教你用ChemDraw 画化学结构式:基础篇
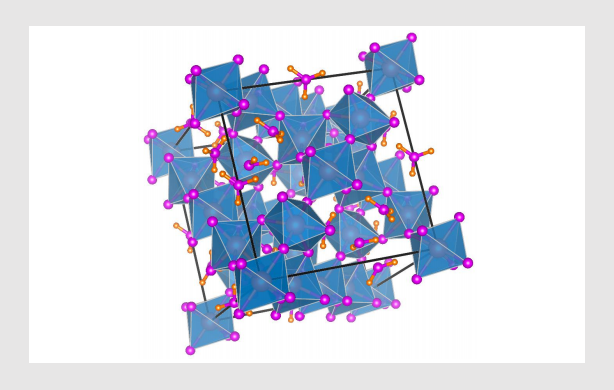
晶体结构可视化软件 VESTA使用教程(下篇)
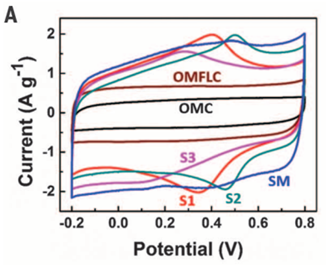
【科研干货】电化学表征:循环伏安法详解(上)
电化学实验基础之电化学工作站篇 (二)三电极和两电极体系的搭建 和测试
【科研干货】电化学表征:循环伏安法详解(下)











