热线:400-152-6858
场发射扫描电镜(SEM)
TEM云现场/现场
SEM云现场/现场
场发射透射电镜(TEM)
更多
X射线衍射仪(XRD)
傅里叶红外光谱仪(FTIR)
电子自旋共振波谱仪(ESR/EPR)
液体核磁共振(NMR)
X射线光电子能谱(XPS)
等离子体光谱仪(ICP-OES)
等离子体质谱仪(ICP-MS)
元素分析仪(EA)
全自动物理吸附仪(BET)
Zeta电位及纳米粒度分析仪(DLS)
常温/高温凝胶色谱(GPC)
差示扫描量热仪(DSC)
热重分析(TG-DTG)
同步热分析仪(TG-DSC)
动态热机械分析仪(DMA)
荧光光谱(PL)
微波网络分析仪/矢量网络分析仪(VNA)
振动样品磁强计(VSM)
电化学分析
聚焦离子束(FIB)
FIB云现场/现场
离子减薄/电解双喷
氩离子抛光(离子研磨)
原位红外(in-situ IR)
原位XPS(In-Situ XPS)
原位XRD
原位微分电化学质谱仪(DEMS)
纳米压痕/纳米(微米)划痕
电子万能试验机
硬度测试
温度冲击试验
盐雾试验
老化试验
37项抗生素检测
激光共聚焦显微镜(CLSM)
负染色技术
生物样本透射电镜(TEM)
生物样SEM
HE染色
CaseViewer(切片全景扫描)
免疫组化(IHC)
免疫荧光(IF)
流式细胞仪(FCM)
细胞毒性/增殖
细胞摄取实验
克隆形成
抗菌性能
菌种鉴定
抗菌培养定制
抗菌防霉技术服务
动物模型
小动物活体成像
皮肤伤口愈合实验
抗肿瘤药物筛选
微生物多样性测序
基因组
转录组
宏基因组
蛋白印迹实验(WB)
SDS-聚丙烯酰胺凝胶电泳
蛋白质鉴定
蛋白表达与纯化
实时荧光定量PCR
ELISA酶联免疫吸附测试
全自动生化检测
血液相容性检测
等温量热滴定仪(ITC)
光学表面等离子共振(SPR)
生物膜层干涉(BLI)
微量差示扫描荧光技术 (nanoDSF)
风味组学全套分析
非靶向代谢组学
靶向定制
短链脂肪酸
蛋白质组学
蛋白甲基化修饰(Methylation)
蛋白质磷酸化修饰(Phosphorylation)
蛋白泛素化修饰(Ubiquitination)
药代动力学
细胞药效筛选
体内药效试验
药物毒理学
二级结构预测
蛋白修饰位点motif分析
蛋白组+代谢组联合分析
单细胞多组学联合分析
单细胞ATAC测序
单细胞免疫组库测序
单细胞转录组测序
医学研究整体方案
农林研究整体方案
农产品检测
药物设计与筛选及验证技术
第一性原理-吸附能
第一性原理-态密度
第一性原理-差分电荷密度
第一性原理-反应能垒
分子动力学-分子对接
生物信息学-网络药理学
分子动力学-电池电解液溶剂化结构
分子动力学-NEMD方法对材料的热导率计算
量子化学-静电势
量子化学-反应路径
量子化学-拉曼光谱
量子化学-吸收谱
固体力学分析
光学-镜头光路模拟
计算流体力学-气动声学
计算流体力学-气动性能计算
相图计算
VASP软件版权
机器学习
生命周期评估(LCA)
机时租用
同步辐射原位表征测试
原位电化学电子自旋共振波谱仪(ESR/EPR)
原位电化学拉曼
三维X射线显微镜XRM(显微CT)
原位扫描电子显微镜(in-situ-SEM)
原位EBSD(in-situ EBSD)
同步辐射吸收谱(XAFS)
同步辐射XAFS数据拟合分析
台式XAFS吸收谱
同步辐射GIWAXS/GISAXS测试
同步辐射XRD
同步辐射全散射PDF
同步辐射散射数据处理
同步辐射X射线断层扫描成像
同步辐射红外测试
同步辐射微区表征测试
论文翻译
降重服务
查重服务
XPS数据分析
XRD数据精修
 显微成像
显微成像 结构分析
结构分析 成分分析
成分分析 物性分析
物性分析 热学性能
热学性能 声光电磁
声光电磁 材料加工
材料加工 原位测试
原位测试 力学性能
力学性能 环境试验
环境试验 显微成像
显微成像 病理检测
病理检测 细胞实验
细胞实验 微生物实验
微生物实验 动物实验
动物实验 测序平台
测序平台 蛋白实验
蛋白实验 分子及生化
分子及生化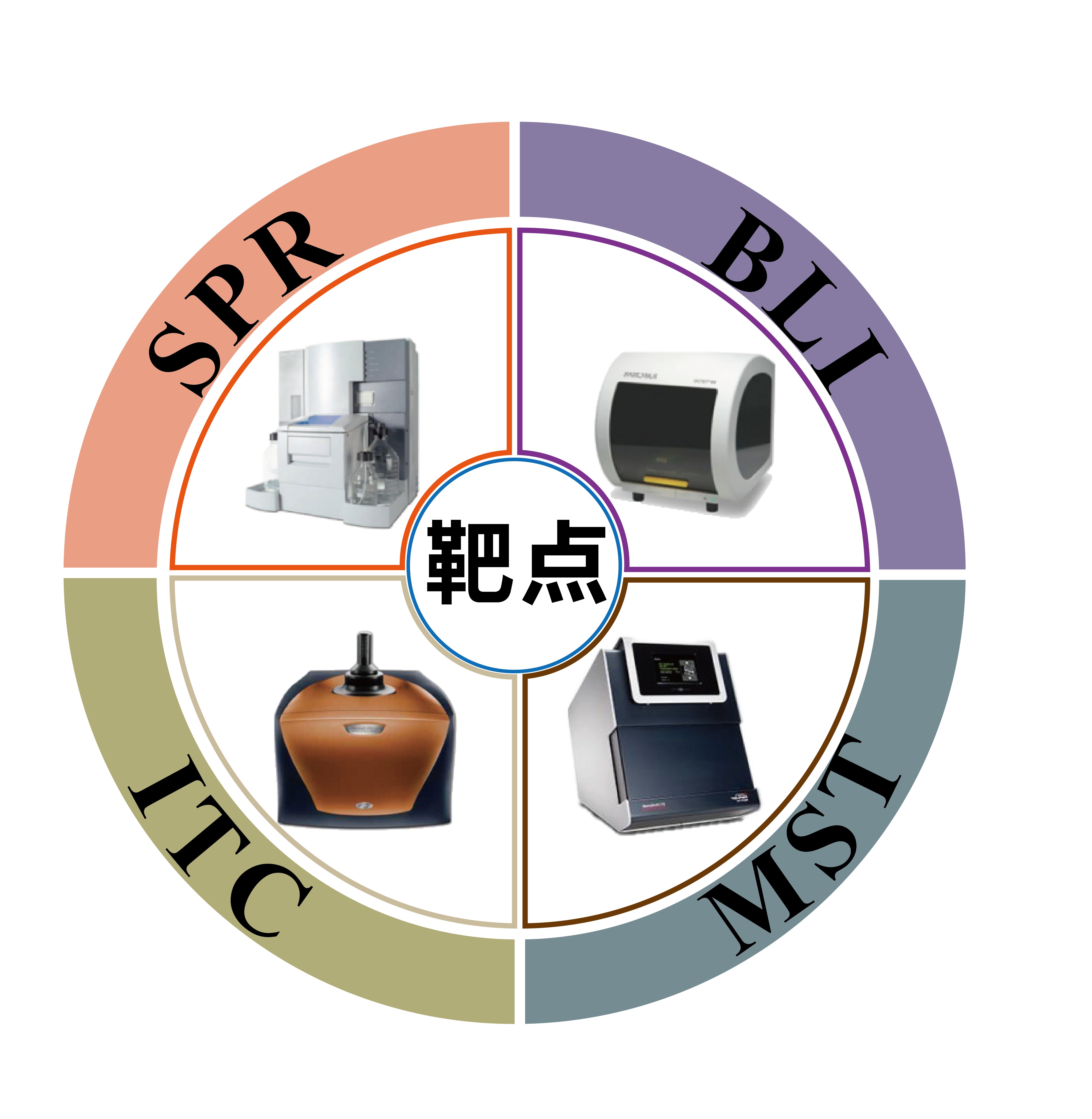 分子互作平台
分子互作平台 代谢组学平台
代谢组学平台 蛋白组学平台
蛋白组学平台 临床前CRO
临床前CRO 生物信息学平台
生物信息学平台 单细胞组学
单细胞组学 整体课题服务
整体课题服务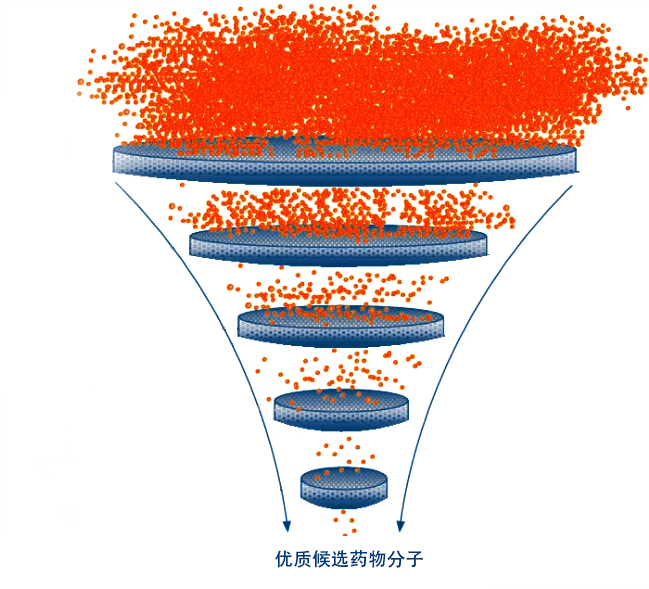 药物筛选平台
药物筛选平台 第一性原理
第一性原理 分子动力学
分子动力学 量子化学
量子化学 CAE仿真
CAE仿真 更多服务
更多服务 超算服务
超算服务 原位变温/气氛/光照测试
原位变温/气氛/光照测试 原位电化学测试
原位电化学测试 原位力学测试
原位力学测试 吸收
吸收 散射/衍射
散射/衍射 原位
原位 成像
成像 光谱
光谱 微区
微区 论文翻译
论文翻译 数据分析
数据分析 基金标书
基金标书 文库百科
文库百科